Spatial gene expression
What is spatial gene expression?
Most cells in our body have functionally specialized regions containing distinct sets of proteins. This complexity relies on the capacity of cells to generate, sustain, and regulate subcellular compartments.
For example, multi-cellular organisms begin from one cell, which lays the spatial blueprint for multi-cellular asymmetry (e.g., left hand, right foot) through localized gene expression.
How the cell achieves such asymmetry is not well understood because of limited experimental approaches for discovering and analyzing localized gene expression. Historical methods – such as microscopy – are generally low-throughput, enabling study of one or two genes at a time, not the 100s - 10,000s of total genes in an organism.
Genome sequencing primer
In the 2000s, genome sequencing had two major applications:
De-novo sequencing of unknown genomes (e.g., strawberry genome, or the cov-2 virus).
Precisely quantifying how much of which genes are expressed under different conditions (e.g., which genes are over-expressed in cancer cells vs normal cells).
Sequencing sub-regions of cells
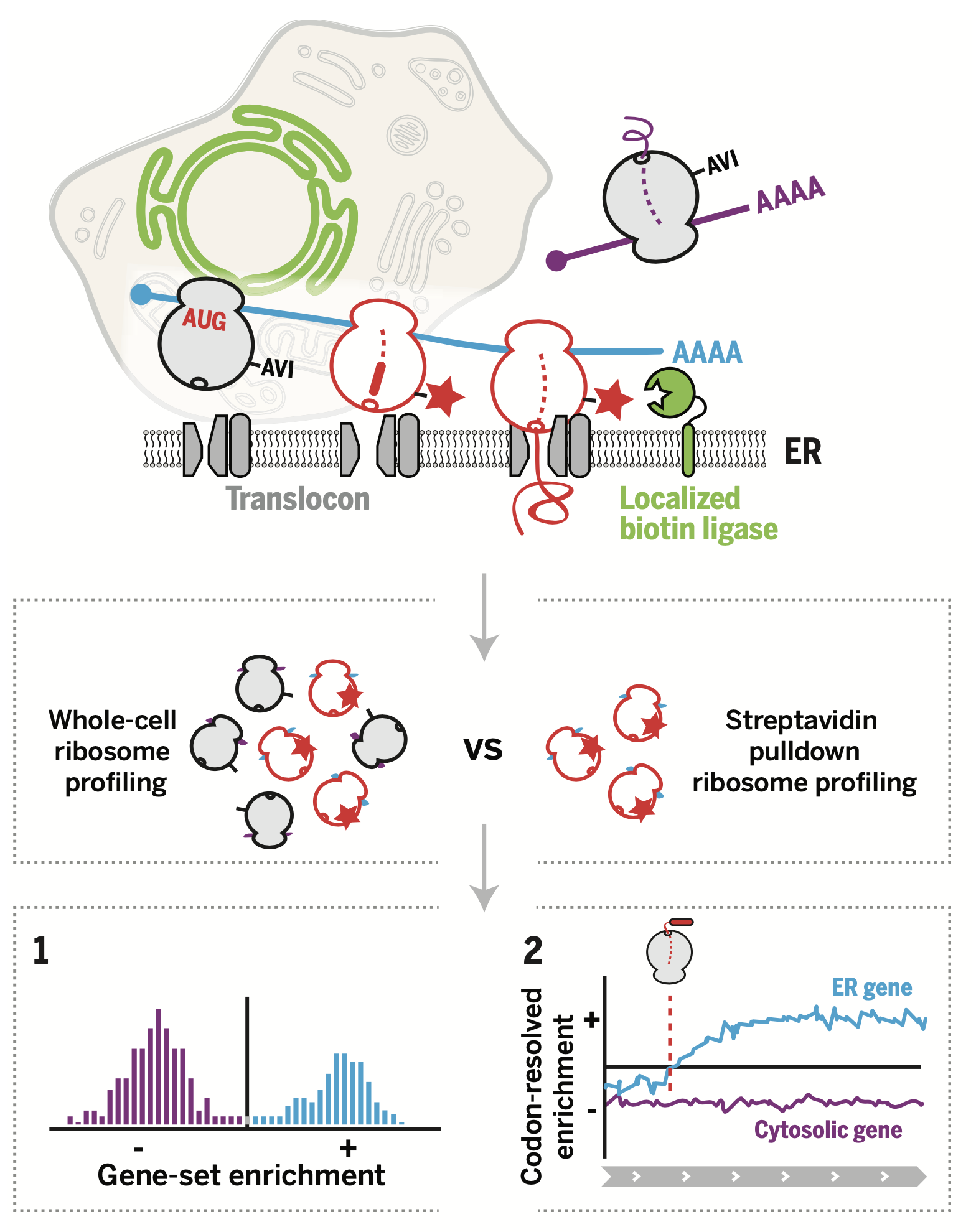
To enable better study of localized gene expression, in grad school I co-developed a genome sequencing approach to enable a global view of localized gene expression within sub-compartments of cells with unprecedented precision.
Our approach was built on a "ribosome profiling" method, previously developed in our lab to quantitatively analyze gene expression (#2 above). We refined this methodology to analyze localized sub-populations of ribosomes, the cellular machines that create proteins, encoded by genes. Our findings may guide the study of region-specific reactions in morphologically complex metazoan organisms.
Still reading?
Check out our papers:
Principles of ER cotranslational translocation revealed by proximity-specific ribosome profiling
Science 346, (2014); DOI: 10.1126/science.1257521
Targeting and plasticity of mitochondrial proteins revealed by proximity-specific ribosome profiling
Science 346, 748 (2014); DOI: 10.1126/science.1257522